為了追求高能量密度的存儲(chǔ)/轉(zhuǎn)換裝置,采用金屬(包括鋰和鈉)作為負(fù)極的可充電電池最近受到關(guān)注。眾所周知的是,即使在低于擴(kuò)散的電流密度下,與金屬負(fù)極接觸的液體電解質(zhì)的化學(xué)穩(wěn)定性,限制了電化學(xué)性能和安全性。其中電解質(zhì)連續(xù)反應(yīng)導(dǎo)致電解液耗盡,界面阻抗增大,此外,電流引起的內(nèi)部短路產(chǎn)生的間接過程也可能導(dǎo)致電池失效。因?yàn)榫哂械墓逃械陌踩阅芎头乐怪L(zhǎng)的功能,固態(tài)電解質(zhì)(SSE)被認(rèn)為是最有前途的方法之一。迄今為止已經(jīng)研究了兩種主要類型的SSE:基于離子導(dǎo)電無機(jī)固體的SSE和基于有機(jī)聚合物的SSE(固態(tài)聚合物電解質(zhì)(SPE))。從各種角度來看,SPE被認(rèn)為具有吸引力,包括其卓越的機(jī)械韌性,低成本,輕質(zhì)特性和與大規(guī)模卷對(duì)卷制備工藝的兼容性。一般來說,SPE應(yīng)該能夠在負(fù)極上實(shí)現(xiàn)高離子導(dǎo)電性和鋰離子的快速界面?zhèn)鬏敚浯危鼞?yīng)該在電池循環(huán)過程中保持機(jī)械穩(wěn)定性和化學(xué)惰性。而在室溫下,大多數(shù)基于在聚(環(huán)氧乙烷)中的LiTFSI的SPE都不能滿足這其中的任何一種要求。
最近,美國(guó)康奈爾大學(xué)Lynden A. Archer教授課題組報(bào)道了在電池中陽離子引發(fā)的分子醚開環(huán)聚合,從而制備固態(tài)聚合物電解質(zhì)(SPEs)的過程。其保持良好的界面接觸,SPEs在室溫下展現(xiàn)了高離子電導(dǎo)率(>1mS/cm),低界面電阻,均勻的鋰沉積和高的鋰沉積/剝離效率(在300次充電-放電循環(huán)后庫(kù)倫效率大于98%)。SPEs在Li-S,Li-LiFePO4和Li-LiNi0.6Mn0.2Co0.2O2等全電池中的成功應(yīng)用,進(jìn)一步證明采用原位設(shè)計(jì)的SPEs可以實(shí)現(xiàn)高的庫(kù)倫效率(>99%)和長(zhǎng)壽命(>700次循環(huán))。此研究為制備滿足實(shí)際需求的固體電解質(zhì)提供了一個(gè)有前景的方向,相關(guān)研究成果以“Solid-
1、固態(tài)聚合物電解質(zhì)(SPE)的制備
在本研究中,通過液體前體的聚合原位合成SPE解決了低離子電導(dǎo)率和高界面電阻的問題。充分利用液體的優(yōu)點(diǎn),包括低粘度和易處理和濕潤(rùn)性,從而與界面產(chǎn)生良好的界面接觸(圖1a)。隨后的開環(huán)聚合反應(yīng)將液體電解質(zhì)從液態(tài)材料轉(zhuǎn)變?yōu)楣虘B(tài)材料,該方法生成了高純度的SPE(與流動(dòng)的液體電解質(zhì)相比),在電解質(zhì)制造或電池組裝時(shí),提供了一種簡(jiǎn)便的方法去修復(fù)在固態(tài)電解質(zhì)產(chǎn)生的界面缺陷。通過執(zhí)行聚合1,3-二氧戊環(huán)(DOL)開環(huán)來評(píng)估這種假設(shè),來制備基于這種材料的固態(tài)電解質(zhì)類似物。在水的存在下,一些有機(jī)鋁化合物,如二乙基氯化鋁和乙基二氯化鋁,引發(fā)DOL的聚合。對(duì)具有相似活性但不需水添加的電解質(zhì)鹽——三氟甲磺酸鋁((Al(CF3SO3)3和Al(OTf)3)鹽是DOL聚合的有效引發(fā)劑,并可用于將基于DOL的液體電解質(zhì)轉(zhuǎn)化為SPE的濃度低至0.5mM。
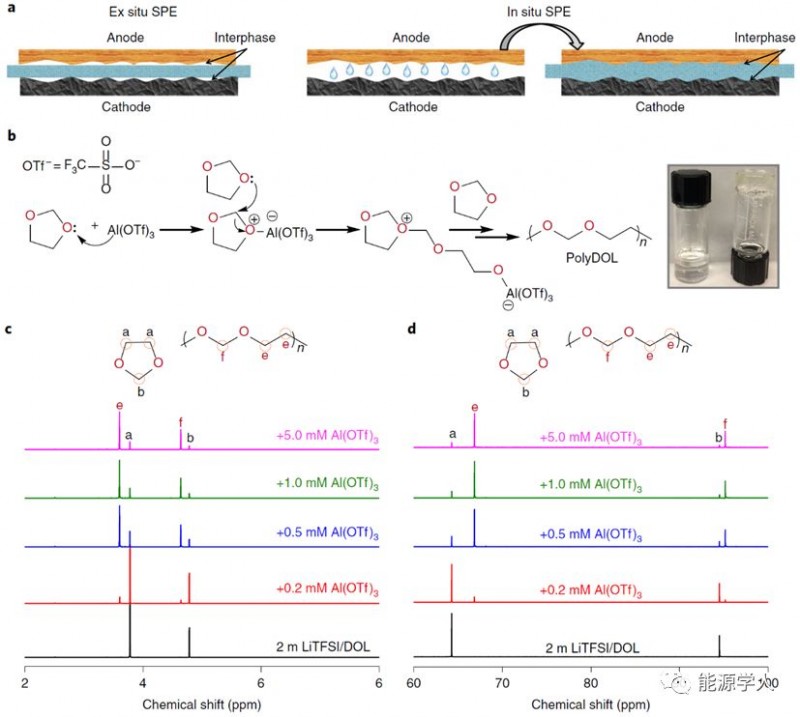
Figure 1. SPEs的制備。a)非原位和原位合成SPEs的示意圖,非原位合成具有更高的界面電阻,原位合成形成的SPEs的液體前驅(qū)體能夠潤(rùn)濕并產(chǎn)生良好的界面接觸;b)Al(OTf)3引發(fā)的DOL聚合的反應(yīng)機(jī)理示意圖,插圖:描繪液體DOL電解質(zhì)(2m LiTFSI/DOL,左)和在含有0.5mM Al(OTf)3鹽的電解質(zhì)中自發(fā)地固態(tài)聚DOL電解質(zhì)的數(shù)碼照片(右);c)液體DOL和在不同Al(OTf)3濃度下形成聚DOL的氫NMR光譜;d)液體DOL和在不同Al(OTf)3濃度下形成聚DOL的碳NMR光譜。
為進(jìn)一步探究SPEs的動(dòng)力學(xué)和電化學(xué)特性,采用差示掃描量熱法(DSC)研究聚DOL的熱轉(zhuǎn)變。圖2a顯示,無鹽聚DOL是晶體,有鹽聚DOL基本上是無定形的,在原位形成的聚DOL電解質(zhì)中結(jié)晶是高離子電導(dǎo)率的關(guān)鍵因素。通過研究聚合過程的動(dòng)力學(xué)彈性和離子傳輸?shù)臅r(shí)間依賴性變化的關(guān)系,來研究電解質(zhì)的性質(zhì)。
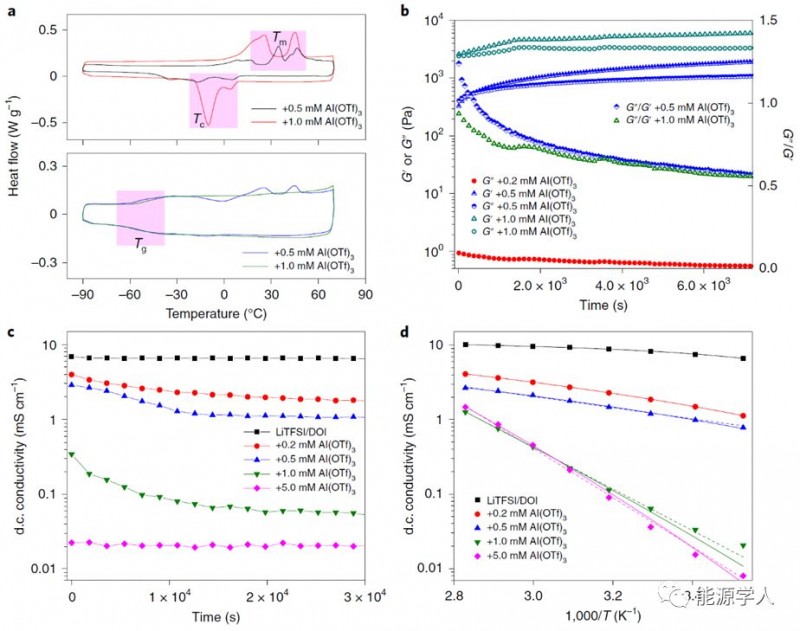
Figure 2. 不同濃度Al(OTf)3的SPEs的動(dòng)力學(xué)和電化學(xué)特性。a)不含LiTFSI(頂部)和含LiTFSI的聚DOL的DSC分析,陰影區(qū)域分別顯示熔融峰(Tm),再結(jié)晶峰(Tc)和玻璃化轉(zhuǎn)變峰(Tg);b)動(dòng)態(tài)存儲(chǔ)(G')和損耗(G'')模量(Pa)和損耗角正切(tanδ=G''/G')與聚DOL電解質(zhì)的時(shí)間的關(guān)系;c)SPE的電導(dǎo)率與聚合時(shí)間的關(guān);d)聚DOL電解質(zhì)的電導(dǎo)率與溫度的關(guān)系。
2、原位形成SPEs的電化學(xué)性質(zhì)
用于制造SPE的前體的DOL-LiTFSI電解質(zhì),由于在高于4V(相對(duì)于Li+/Li)電位下相對(duì)差的氧化穩(wěn)定性,因此不常用于采用嵌入正極的鋰電池中,圖3a中的結(jié)果清楚地表明,聚合反應(yīng)可以將氧化穩(wěn)定性提高到5 V以上。圖3b中顯示當(dāng)電壓低于4.7 V時(shí),在SPE中測(cè)量的漏電流很小(<20uA)。相比之下,使用液體LiTFSI/DOL電解液時(shí),當(dāng)電壓低于4.3V時(shí),漏電流超過1mA。具有0.5m MAl3+的SPE表現(xiàn)出最高機(jī)械模量和室溫離子電導(dǎo)率,配比SPE的Li-Cu電池表現(xiàn)一致且高庫(kù)侖效率(>98%),即使在300 次循環(huán)后也是如此。同時(shí),使用原位形成的聚DOL SPE和未聚合液體電解質(zhì)在對(duì)稱電池中研究鋰的沉積與剝離(圖3e)。

Figure 3. 聚DOL電解質(zhì)的電化學(xué)穩(wěn)定性。a)液體DOL(2m LiTFSI/DOL)電解質(zhì)和聚DOL SPEs(0.5mM Al(OTf)3+2m LiTFSI/ DOL)的線性掃描伏安,掃描速率為1mV/s。插圖:以1mV/s的掃描速度,鋰在聚DOL SPEs中沉積-剝離的電流-電壓曲線;b)使用NMC作為正極,在液體DOL電解質(zhì)和聚DOL SPEs中的電化學(xué)浮動(dòng)分析;c)在聚DOL SPEs中,鋰沉積-剝離與循環(huán)圈數(shù)的關(guān)系曲線;d)當(dāng)電流密度為1mA/cm2,鋰沉積容量為1mAh/cm2時(shí),在液體DOL電解質(zhì)和聚DOL SPE中,相應(yīng)的庫(kù)侖效率與循環(huán)圈數(shù)的關(guān)系曲線;e)當(dāng)電流密度為1mA/cm2,鋰沉積容量為1mAh/cm2時(shí),使用液體DOL電解質(zhì)和聚DOL SPE對(duì)稱電池的長(zhǎng)循環(huán)性能。
3、SEI膜與鋰的電化學(xué)沉積
進(jìn)一步研究了鋰金屬負(fù)極在聚DOL電解質(zhì)中SEI的組分和化學(xué)狀態(tài)。在液體DOL電解質(zhì)中,鋰片循環(huán)50圈之后呈現(xiàn)獨(dú)特的黑色,并有明顯的裂縫(圖4a);而在基于聚DOL電解質(zhì),鋰片表面沒有明顯的變化(圖4c)。使用X射線光電子能譜(XPS)光譜用于表征兩個(gè)系統(tǒng)中SEI的化學(xué)狀態(tài),聚DOL SPE可以在其表面產(chǎn)生大量的LiF,而且鋁源往往會(huì)緩慢地移動(dòng)到它們之間的界面,DOL聚合過程也可以通過還原鋰負(fù)極表面的DOL。原位形成的SPE的優(yōu)點(diǎn)是,大多數(shù)的電解質(zhì)溶劑是聚合的,而不僅僅是相對(duì)薄的電極表面的層。
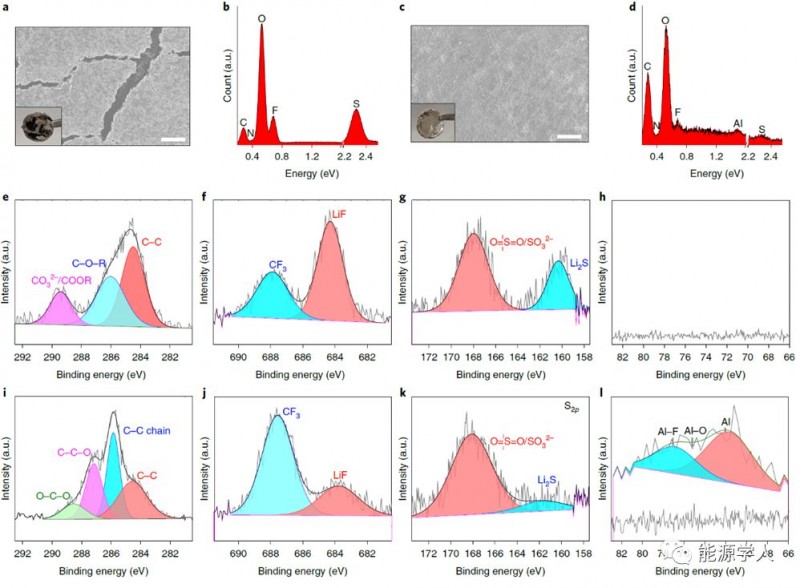
Figure 4. 在液體DOL和SPE電解質(zhì)中鋰循環(huán)形成SEI的表征。a, b)在液體電解質(zhì)中,對(duì)稱電池循環(huán)50圈之后鋰片的SEM圖像和EDS分析;c, d)在在聚DOL SPEs中,對(duì)稱電池循環(huán)50圈之后鋰片的SEM圖像和EDS分析;e-l)相對(duì)應(yīng)的XPS分析,其中(e-h)在液體電解質(zhì)中循環(huán),(i-l)在聚DOL SPEs中循環(huán)。
進(jìn)一步使用可視化系統(tǒng)研究在兩種電解質(zhì)系統(tǒng)中的形貌演變,對(duì)于使用液體電解質(zhì)的對(duì)稱電池,鋰沉積過程中,能清晰看見苔蘚狀的鋰,而在聚DOL SPE中,鋰沉積層更加緊密,聚DOL SPE的彈性在循環(huán)期間提供了彎曲和伸展的機(jī)制,以適應(yīng)電沉積期間的體積變化。此外,DOL聚合形成的SPE增強(qiáng)了DOL接觸時(shí)的化學(xué)穩(wěn)定性,對(duì)金屬表面的副反應(yīng)被壓制了。這種功能的組合,包括有機(jī)彈性聚合物,鋁配合物和富含LiF的無機(jī)化合物被認(rèn)為是SPE具有優(yōu)異界面屬性的原因。

Figure 5. 在液體電解質(zhì)和聚DOL SPE中的鋰電沉積形貌。a, b)在液體電解質(zhì)中,鋰沉積的實(shí)時(shí)光學(xué)顯微鏡形貌演化表征(a)和相對(duì)應(yīng)的光學(xué)照片和SEM圖像(b);c, d)在聚DOL SPE中,鋰沉積的實(shí)時(shí)光學(xué)顯微鏡形貌演化表征(c)和相對(duì)應(yīng)的光學(xué)照片和SEM圖像(d)。沉積電流密度為2mA/cm2,測(cè)試持續(xù)60分鐘。
4、原位形成SPEs的全電池性能
為了證明原位形成的SPE的優(yōu)點(diǎn),與各種正極材料匹配組成可充電鋰金屬電池。與硫匹配的Li-S電池中,多硫化鋰的溶解和電解質(zhì)與鋰的副反應(yīng)是主要問題,在SPE中不使用添加劑LiNO3可以有效穩(wěn)定Li-S電池的根本的原因是它會(huì)抑制多硫化物的溶解。如圖6a所示,與DOL電解液相比,使用聚DOL SPE和CMK-3/硫復(fù)合正極的Li-S電池可以在更寬的電壓范圍運(yùn)行且擁有接近100%的高庫(kù)侖效率。通常情況下,電解質(zhì)能夠滲透到多孔正極結(jié)構(gòu)中是具有良好離子傳輸?shù)囊螅褂肗MC(622)和LFP正極與之匹配,聚DOL SPE電池顯示出高庫(kù)侖效率和出色的可逆性。

Figure 6. 使用鋰金屬負(fù)極和聚DOL SPEs的全電池電化學(xué)性能。a)在0.1C的倍率下,Li/聚DOL SPE /S電池的充放電曲線;b)在0.1C的倍率下,Li/聚DOL SPE/NMC(622)電池的充放電曲線;c)在0.1C的倍率下,Li/聚DOL SPE/LFP電池的充放電曲線;d)在Li//DOL//LFP(紅色)和Li / 聚DOL SPE/LFP(藍(lán)色和紫色)中的恒電流循環(huán)性能和庫(kù)倫效率。